

深度揭示煤體中多相水分(吸附相與自由相)的分布規律對煤層氣工程開發至關重要,然而其定量表征方法與微觀機理研究仍存在空白。本文通過理論與實驗相結合,提出了一種創新且更精確的煤體多相水表征方法,該方法不僅適用于飽和含水煤體,也適用于未飽和狀態。首先,對六種不同變質程度的煤樣開展傳統核磁共振(NMR)-離心聯測實驗,以評估實驗多相水分布特征。結果表明,自由水含量隨離心壓力增加顯著上升,最終趨于穩定極限值,該變化規律近似符合類Langmuir方程,可進一步用于估算理論多相水含量。此外,受實驗室離心機壓力極限所限,實驗測得的吸附水含量普遍高于理論值。通過整合離心實驗數據與典型孔隙結構參數,結合吸附比例方程,建立了改進型多相水分析模型。研究顯示,吸附水密度均值為1.59 g/cm3,平均吸附層厚度為0.63 nm。煤體表面弛豫率最優估值分布于2.2-5.2 nm/ms區間。實驗估算的多相水核磁共振譜圖與理論模型存在系統性偏差,表明僅依靠實驗數據不僅會導致實驗室分析誤差,更將影響礦場測井解釋的準確性。本文提出的多相水評估新方法可實現(未)飽和煤體中吸附水/自由水含量及空間分布的定量表征。
煤層氣開發在化石清潔能源補充、降低煤礦安全事故及減少溫室氣體排放方面具有關鍵作用。煤層氣儲層在原狀地層環境中呈現為液-氣-固三相共存體系。其中,作為不可忽略的流體組分,煤孔隙-裂隙系統中水分的動態變化會改變儲層物性特征,進而影響煤層氣水力壓裂效果。煤中水分可分為性質迥異的兩種相態:其一為吸附相水,其二為自由相水。
孔隙-裂隙系統中賦存的多相水(吸附相與自由相)深刻影響甲烷的吸附/解吸-運移-產出的全過程,其動態作用機制可歸納為:(a)孔隙水的存在會占據形態學空間區域,導致原位條件下游離甲烷含量顯著降低;(b)由于競爭吸附效應,吸附水分子會占據基質表面的甲烷吸附位點,直接導致甲烷吸附容量下降;(c)此外,煤孔隙-裂隙系統中的水鎖效應會阻礙甲烷與基質表面接觸,進一步削弱甲烷吸附能力;(d)孔隙水對甲烷運移產生顯著阻遏作用,進而影響甲烷擴散能力與滲透率;(e)煤層氣產能受甲烷賦存數量與運移特性的顯著制約,而這些特性與煤中多相水分布密切關聯。因此,準確評估煤中多相水特征對煤層氣勘探開發具有重要意義。
盡管學界已從多角度對煤中多相水特征展開研究,但由于缺乏精確評估吸附相與自由相水行為的理論與實驗方法,煤孔隙內液態水的微觀作用機制仍不明確。本文提出了一種結合核磁共振-離心實驗與液體吸附理論的煤多相水定量表征新方法。全文研究框架如下:首先,對六種不同變質程度煤樣開展系列核磁共振-離心測試,逐級估算多相水分布;其次,通過整合核磁共振-離心實驗與低溫氮吸附實驗數據,建立改進的吸附比例模型;繼而,系統確定完全飽和狀態下理論多相水分布特征;最后,探討本研究提出的多相水定量表征方法在現場評估中的應用前景。
本研究采集了來自中國準噶爾盆地、鄂爾多斯盆地和沁水盆地的六種不同煤級煤樣。基于鏡質組最大反射率指標,所選煤樣涵蓋次煙煤、高揮發分煙煤、中揮發分煙煤、低揮發分煙煤、貧煤及無煙煤(表1)。
表1 實驗煤樣詳細巖石物理特征
樣品制備過程中,將大塊煤樣破碎至60-80目(0.25-0.18 mm),隨后置于368 K(95 ℃)烘箱中干燥12小時以去除雜質氣體與水分。樣品干燥后,將所有煤粉置于真空系統中脫氣5小時以確保充分去除殘留氣體。隨后在77 K(-196 ℃)條件下測試氮氣吸附-脫附等溫線以獲取孔隙結構信息。
核磁共振-離心聯測實驗流程如下(圖1):(1)沿煤層層面方向鉆取直徑2.5 cm、長度5.0 cm的圓柱狀煤芯;(2)將煤芯置于368 K真空干燥箱中處理12 h;(3)將干燥煤樣移入紐邁核磁共振儀(型號:中尺寸核磁共振成像分析儀MesoMR23-060H,蘇州紐邁分析儀器股份有限公司 )采集基礎核磁信號;(4)將干燥煤芯置于真空飽和裝置中,采用去離子水在壓力環境下飽和處理至少24 h;(5)用無磁性保鮮膜包裹飽和水煤樣,置入核磁共振儀測定T2分布;(6)將煤芯分別置于1.51、2.17、2.95、3.86、4.88、6.03及7.29 MPa離心壓力(對應離心機11000轉/分鐘工況)下處理2 h以達到理想離心狀態;(7)采用同一臺紐邁核磁共振儀測定不同離心壓力處理后煤樣的T2分布。需說明的是,本實驗采用的核磁共振參數與前期研究保持一致。

圖1 核磁共振-離心實驗流程示意圖
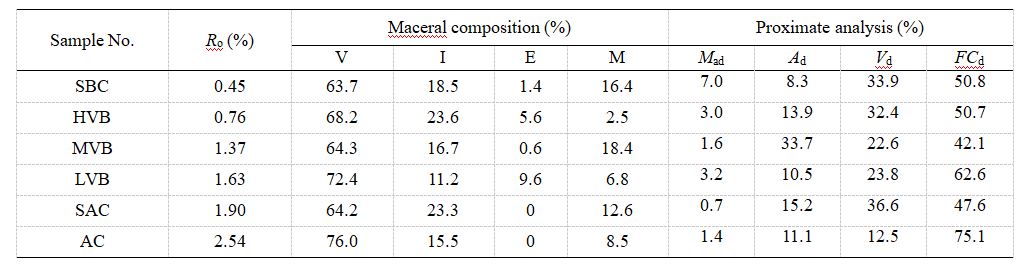
純水的T2譜呈單峰分布,峰值位于200-1000 ms區間。值得注意的是,隨著水質純度提升,T2譜峰呈現左移現象,這可能是由于更高純度水中氫核的自旋運動受到抑制所致。此外,純水的核磁共振總信號幅度與其質量呈線性相關(相關系數約0.9933),表明可直接通過核磁信號幅度計算樣品含水量。
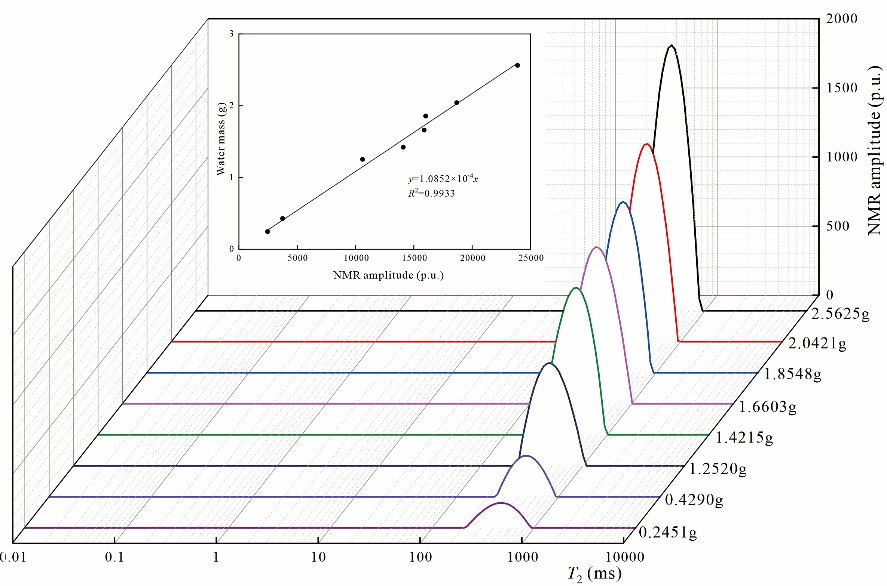
圖2 不同水質純水的T2分布特征
離心實驗是評估煤中多相水特征的常用方法,其理論依據在于自由水會被離心驅替,而吸附水則保留在孔隙系統中。飽和水煤樣的核磁共振T2譜呈現多峰分布特征:最左側峰(峰1)位于0.01-10 ms區間,中間峰(峰2)分布于10-100 ms范圍(圖3)。對于SBC等特定煤樣,在100-1000 ms區間可觀測到第三個譜峰(峰3),表明該類煤樣發育裂隙結構。
隨著離心實驗的進行,不同譜峰的T2分布呈現差異化演變規律。峰1的核磁信號幅度隨離心壓力增加僅略有降低(圖3),而峰2與峰3的信號幅度則呈現顯著衰減趨勢(圖3)。這種信號衰減是由于自由水被離心驅替所致。
圖3 不同離心條件下煤樣的T2譜
1.3 基于低溫氮氣吸附的孔隙結構特征
低溫氮氣吸附實驗測得的BET比表面積與BJH孔體積結果列于表2,其值分別分布于0.51–3.84 m2/g與0.63–10.76×10-3 mL/g區間。圖4展示了約77 K溫度下的氮氣吸附/脫附曲線,根據滯后環形態差異可細分為四種類型(表2):(1)如SBC樣品所示,在P/P?=0.4–1區間存在明顯吸附/脫附滯后環,表征墨水瓶形孔隙結構;(2)如AC樣品所示,吸附支與脫附支近乎重合且滯后環可忽略,主要對應半開放板狀孔隙;(3)如HVB樣品所示,曲線形態與AC樣品類似,但在P/P?=0.7–1區間存在微弱滯后環,指示開放板狀孔隙特征;(4)如MVB樣品所示,除存在明顯滯后環外與SBC樣品曲線相似,反映半開放錐形孔隙結構。
表2 實驗煤樣低溫氮氣吸附測試結果
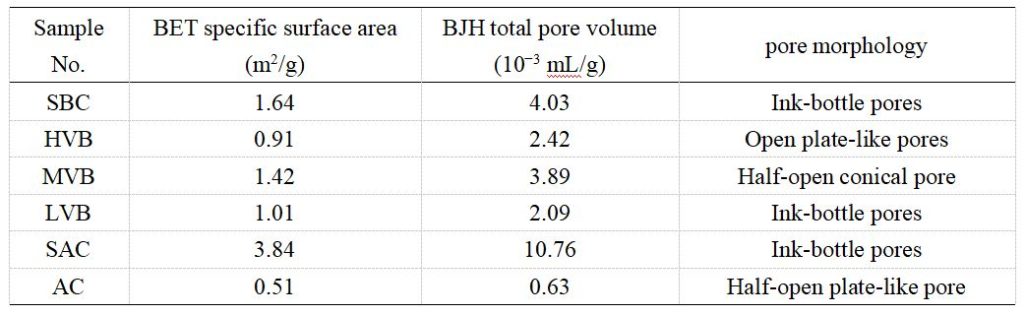
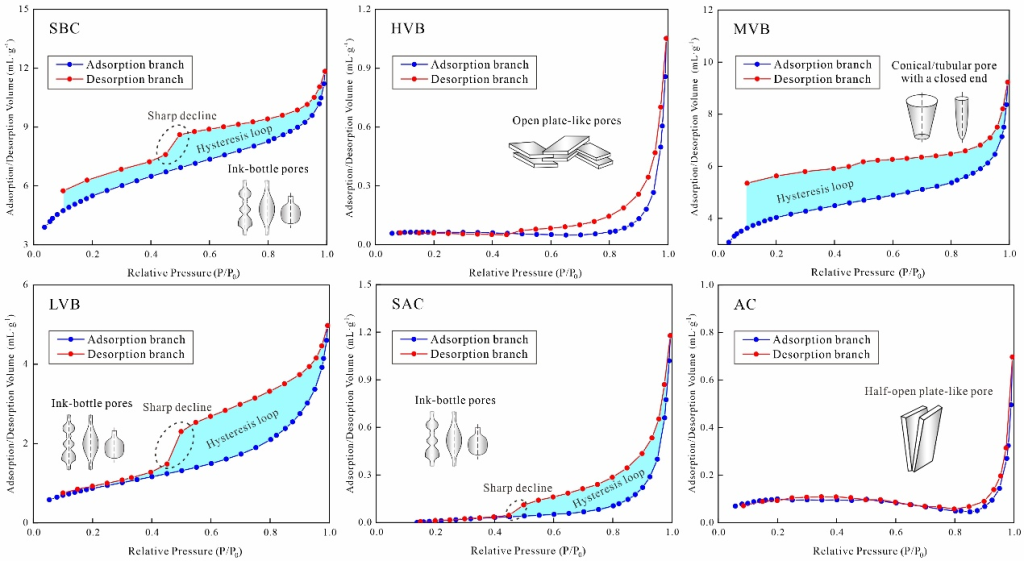
圖4 煤樣的低溫氮氣吸附/脫附曲線
離心過程中,自由相水被驅替排出,而保留于原始孔隙系統的水相則表征為吸附相。通常優選離心設備的最大離心壓力進行多相水含量評估——以最大限度去除自由水。
圖5展示了根據飽和水狀態與最大離心壓力下核磁共振T2譜確定的煤樣多相水分類。經最大壓力離心后,最右側兩個譜峰的信號幅度顯著降低。通過與左側T2譜峰對比可知,自由相水分布于消失區域(圖5綠色區域),而最大離心壓力后的T2譜則表征吸附相水。基于核磁信號幅度與水質量的定量關系(見1.1節),通過核磁共振-離心實驗測定的自由水與吸附水含量如圖6所示。結果表明:實驗測定的自由水含量分布于4.26-24.82 mg/g區間,平均值為15.44 mg/g;吸附水含量介于15.89-39.27 mg/g,平均值為25.20 mg/g。吸附相權重比為0.16-1.05,總體以吸附相水為主導。
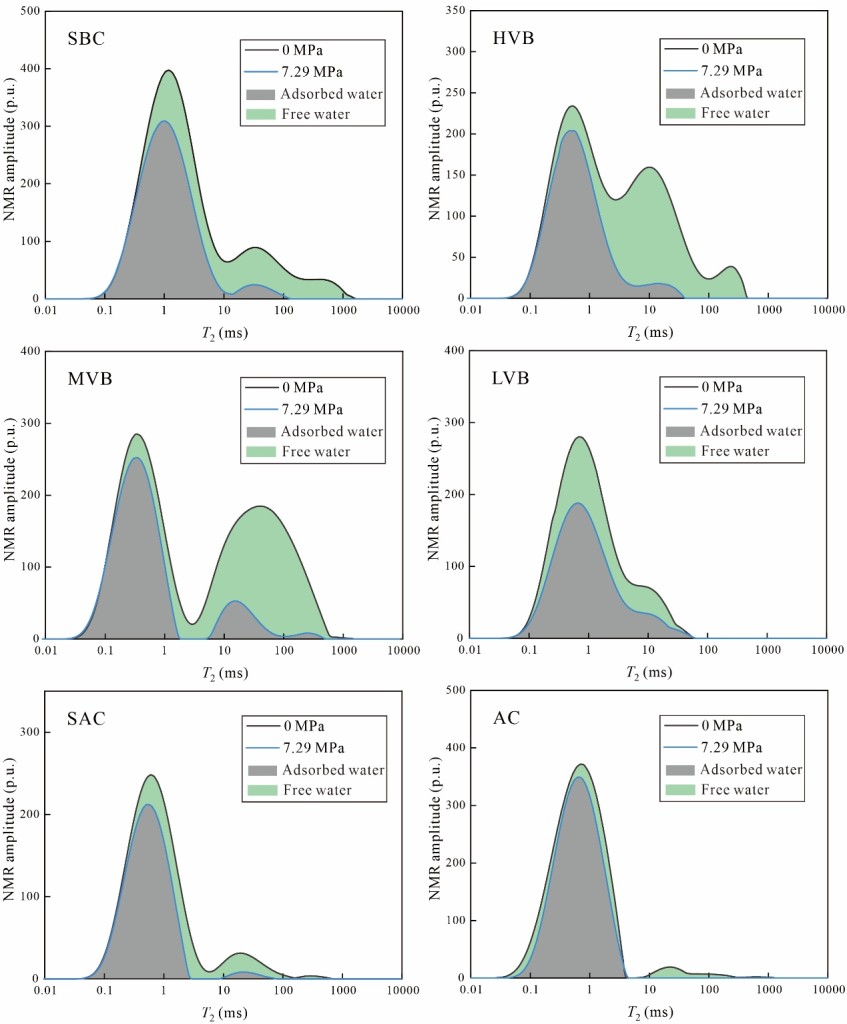
圖5 基于核磁共振-離心實驗方法的多相水分類
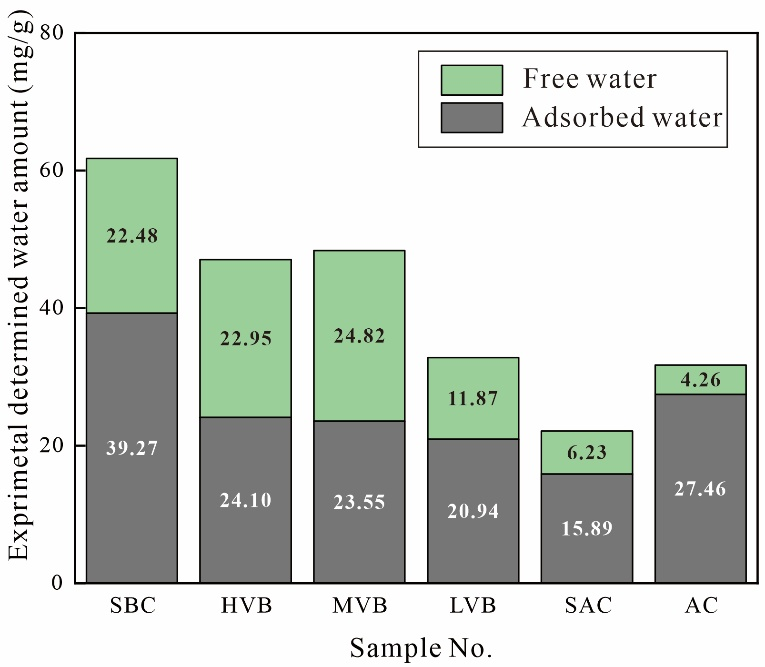
圖6 實驗測定的煤樣自由水與吸附水含量
如圖7和表2所示,可動水含量隨離心壓力的變化規律顯示:特定煤樣的自由水含量隨離心壓力增加而上升,其變化趨勢符合朗繆爾方程擬合曲線(相關系數0.9769-0.9950,表3)。因此,特定離心壓力下的可動水含量可用類Langmuir方程描述:

其中,Cm代表實驗測試獲得的可動水含量,單位為mg/g;Cf代表無限大離心壓力下的最大可動水含量(即自由水含量),單位為mg/g;ΔP代表實驗離心壓力,單位為MPa;ΔPL代表中值離心壓力,單位為MPa。
表3 實驗煤樣巖石物理特征參數
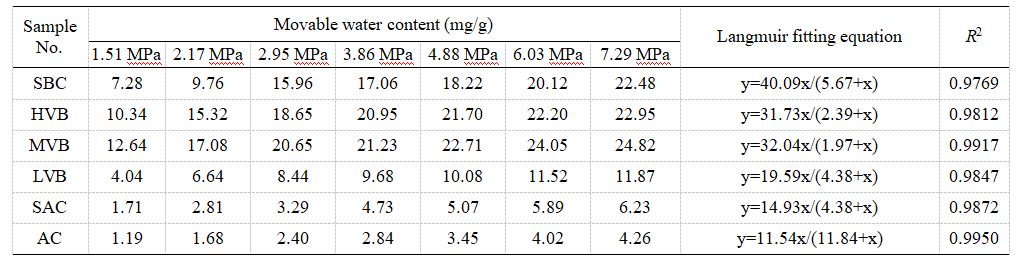
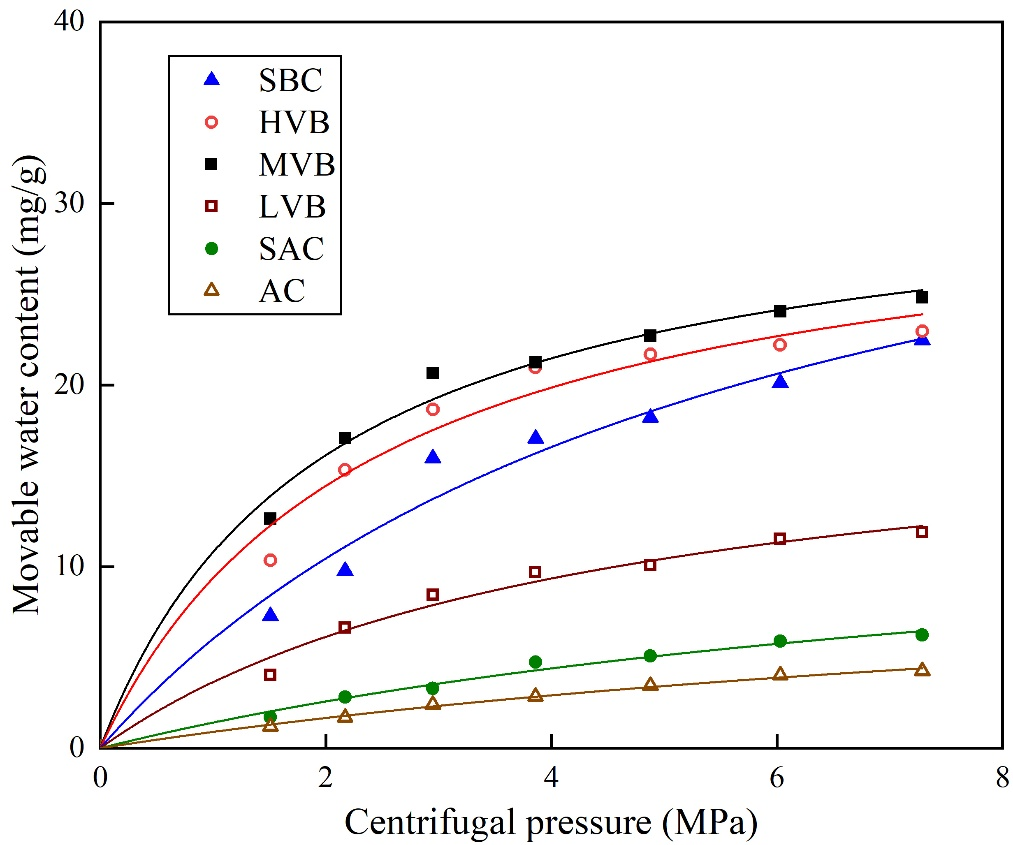
圖7 煤樣離心壓力與可動水含量關系
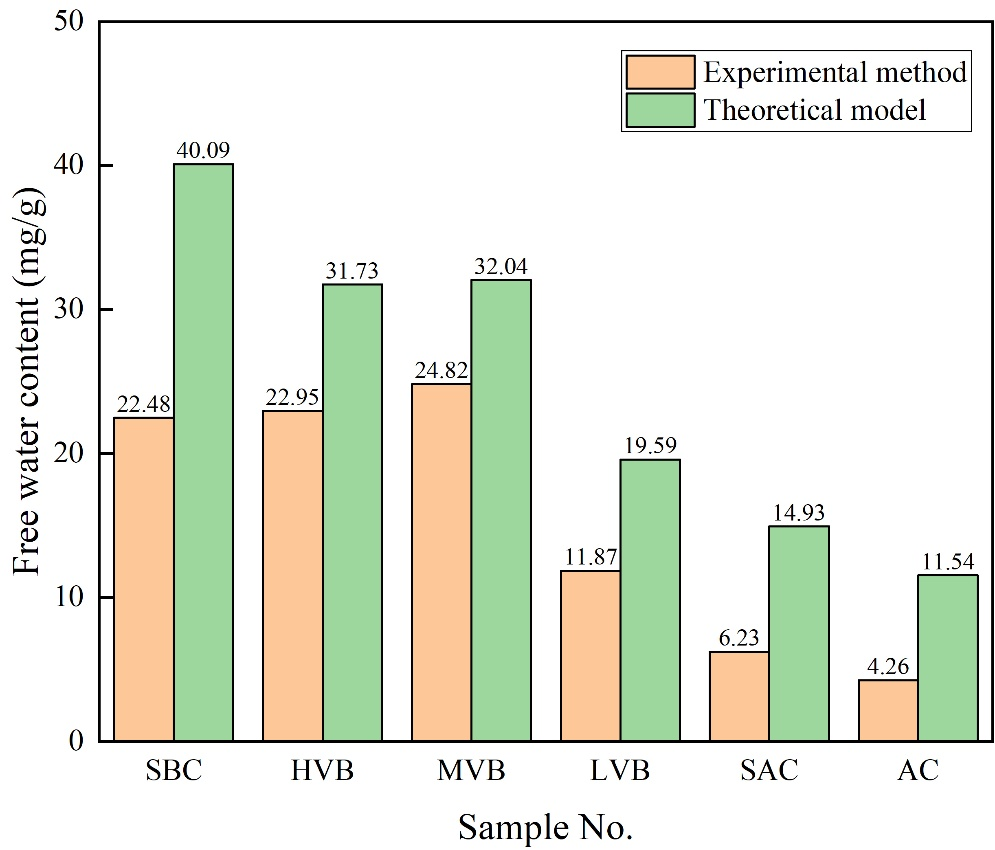
圖8 離心實驗法與理論模型測定的自由水含量對比
微觀尺度上,正如分子模擬所驗證,水分子以有序方式吸附于孔隙表面形成吸附層,同時以自由態分布于孔隙內部。Li等學者提出的吸附比例方程理論模型,可進一步用于表征真實多相水分布。該吸附比例方程可簡表述為:

其中,ra為吸附水與總水的質量比,量綱為1;Ca為理論計算的煤樣吸附水含量,單位為mg/g;Cf為理論計算的煤樣自由水含量,單位為mg/g;ρa與ρf 分別為吸附水與自由水的密度,單位為g/cm3;V為總孔體積,單位為10-3 mL/g;S為比表面積,單位為m2/g;H為平均吸附層厚度,單位為nm。(注:S與V參數分別通過BET理論和BJH模型從低溫氮氣吸附數據獲取。)
基于核磁共振測量原理,對應水分子的T2弛豫時間可表示為:

其中,ρ2為煤體表面弛豫率,單位為nm/ms。聯立公式(2)與(3),可推導出理論吸附比例的表達式為:

實驗測定吸附比例與公式(4)理論值之間的差異——無限趨近于零——標志著表面弛豫率達到最優值。獲得各煤樣表面弛豫率后,按如下方法估算每個T2時間點(T2i)的吸附比例:

需說明的是,核磁共振-離心實驗在室溫(約25℃)條件下進行,因此標準大氣壓下的自由相水密度(ρf)取0.997 g/cm3。如圖9所示,體積-表面積比與自由相-吸附相水含量比值呈現顯著正線性關系。基于圖9擬合結果與公式(2),可計算出吸附相水密度ρa為1.59 g/cm3,平均吸附層厚度H為0.63 nm。
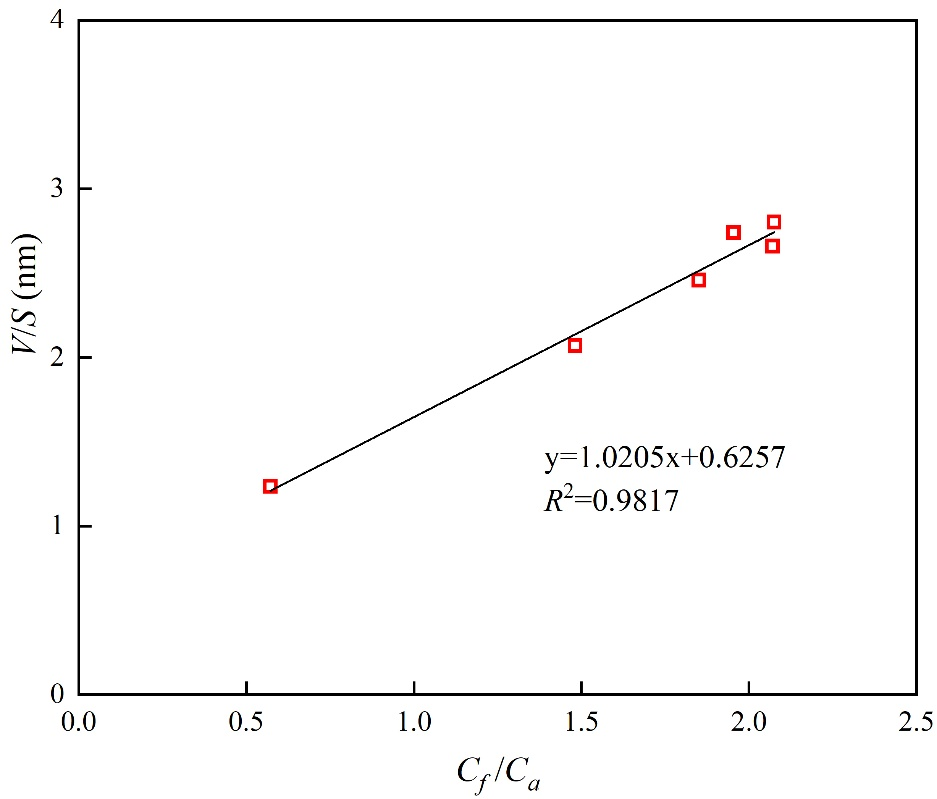
圖9 煤樣中自由/吸附水含量比(Cf/Ca)與孔體積/比表面積比(V/S)關系
基于實驗吸附比例(取值見第4.1.1節)無限趨近于理論值的假設,煤體表面弛豫率可通過下式進行計算:

通過賦予不同表面弛豫率數值,圖10展示了實驗吸附比例與理論值的絕對誤差δ變化規律。模擬結果表明:絕對誤差隨表面弛豫率增大呈現先減小后增大的趨勢,且在誤差趨近于零時存在最優解。經計算獲得煤樣的最優表面弛豫率分布于2.2-5.2 nm/ms區間,平均值為3.35 nm/ms。
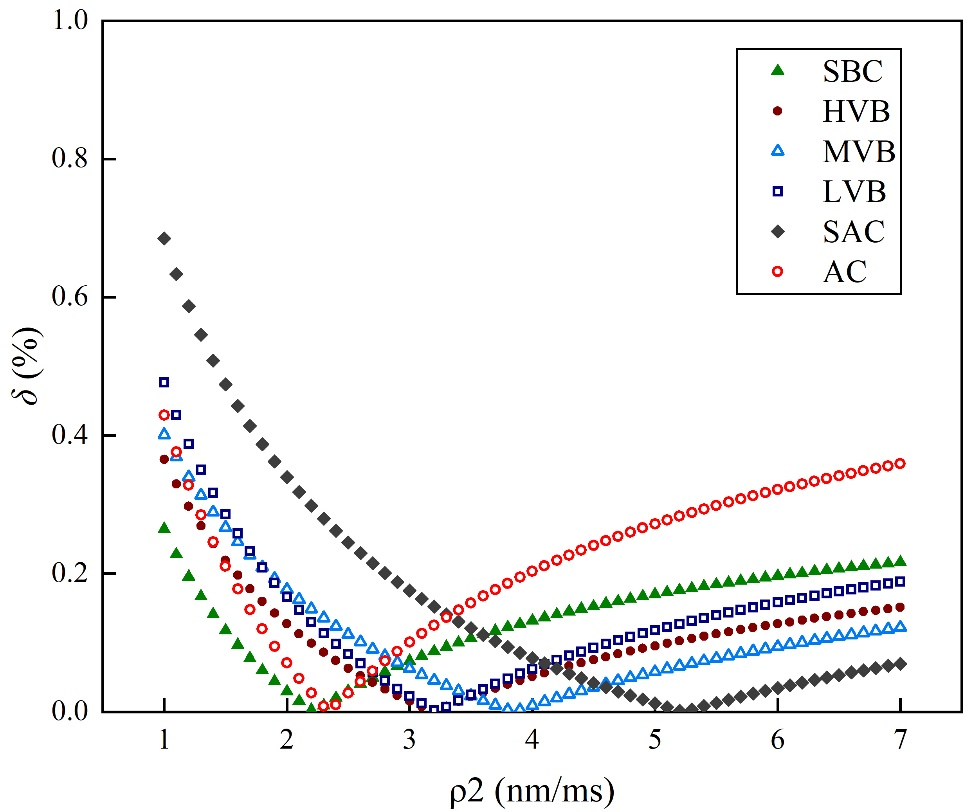
圖10 煤樣最優表面弛豫率計算
基于公式(5)的吸附比例模型及完全飽和狀態T2分布數據,吸附相與自由相水的核磁共振T2特征如圖11所示。結果表明:吸附相水主要分布于較小T2區間(約0.02-10 ms),且呈單峰分布特征;自由相水則對應較長橫向弛豫時間,呈現多峰分布結構。值得注意的是,煤體中吸附相與自由相水的T2分布存在重疊區域,表明在特定孔徑范圍內多相水必然共存。
理論上存在臨界孔徑值(記為rc):當孔隙半徑小于rc時僅存在吸附相水,而當孔隙半徑大于rc時吸附水與自由水共存。根據核磁共振理論方法,孔隙半徑(r)與T2弛豫時間的關系可表述為:

其中,Fs為孔隙幾何因子:平行板狀孔隙取值為1,柱狀孔隙取值為2,球狀孔隙取值為3。結合低溫氮氣吸附測定的孔隙形態與煤體表面弛豫率,圖12展示了吸附/自由相水質量比與孔徑的關聯規律。結果表明:吸附相水隨孔徑增大而逐漸減少,自由相水則呈現持續增加趨勢。平行板狀、柱狀與球狀孔隙的臨界孔徑閾值分別為H、2H與3H。當平行板狀、柱狀與球狀孔隙尺寸分別小于0.63 nm、1.26 nm與1.89 nm時,孔隙水完全由吸附相主導。
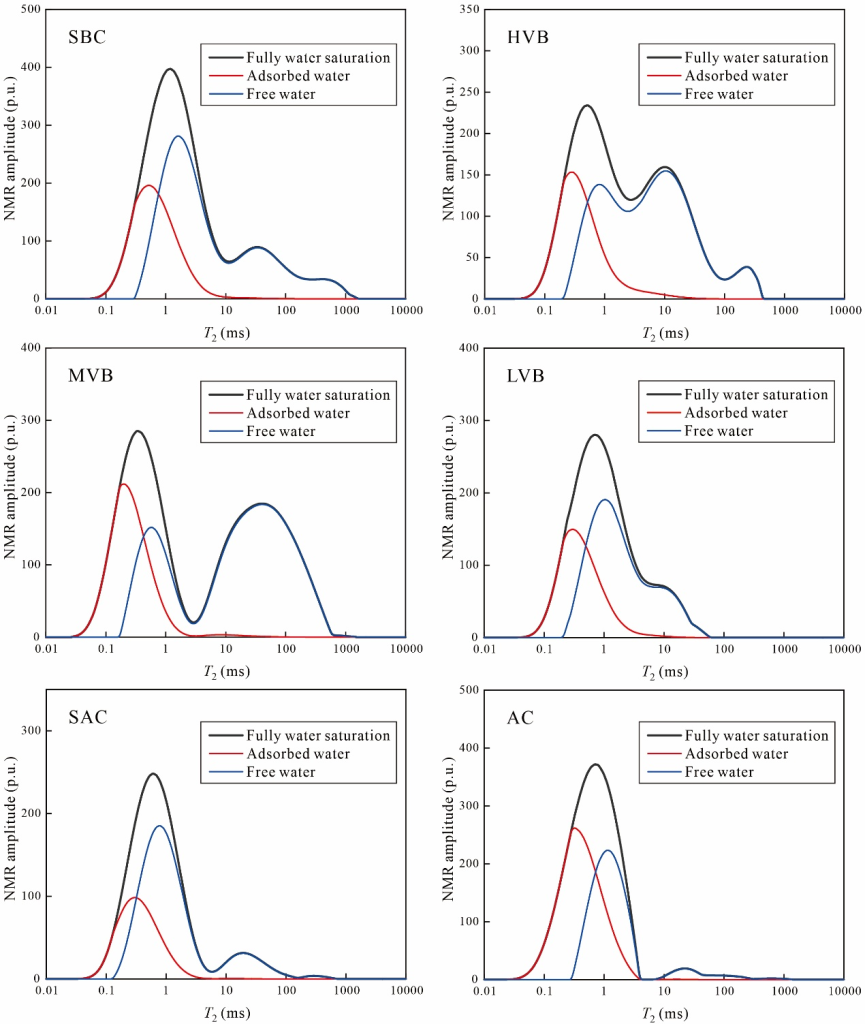
圖 11 煤樣中吸附相與自由相水的理論核磁共振T2分布特征
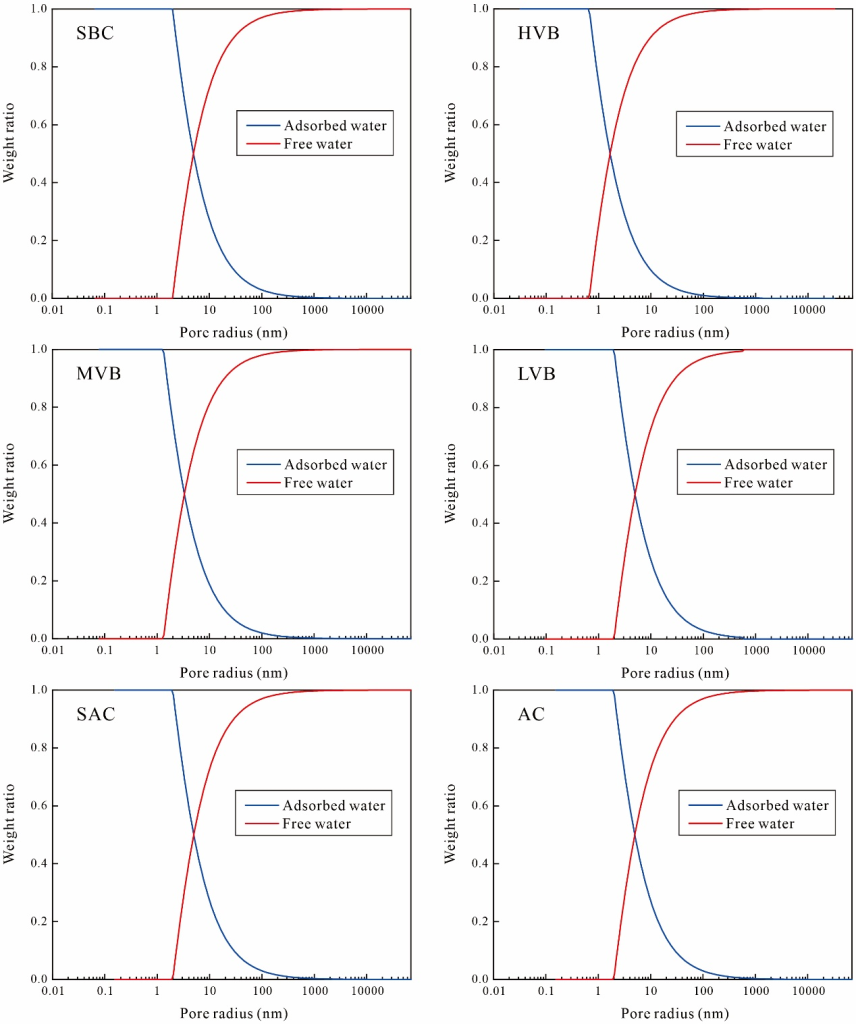
圖?12 吸附/自由相水質量比與孔徑的對應關系
現場煤體多相水含量通常基于核磁共振測井與經驗性T2截止值(T2C)聯合評估,遵循以下原則:T2 < T2C對應吸附相水,T2 > T2C對應自由相水。但需注意,不同物化性質的煤樣其T2截止值存在顯著差異,直接應用核磁共振測井可能導致現場多相水含量評估失真。本文建立的吸附比例方程多相水再評估方法,不僅適用于實驗室分析,還可擴展至現場多相水評估。
該方法的現場實施流程如下:(a)將新鮮煤芯置于無磁性密封樣品罐以保持原始含水狀態;(b)將樣品罐置于核磁共振儀測定原始T2分布;(c)取出煤芯轉入真空飽和裝置實現完全水飽和;(d)通過低溫氮氣吸附測試獲取比表面積與孔體積;(e)基于最大相似性理論(即公式(6))計算最優核磁共振表面弛豫率;(f)結合步驟(e)與公式(5)建立吸附比例模型(公式(5)中吸附水相密度與平均吸附層厚度默認取1.59 g/cm3與0.63 nm);(g)融合原始T2分布(步驟a)與吸附比例分析結果(步驟f)評估吸附/自由水核磁共振分布。
本研究基于核磁共振測試與吸附比例方程,建立了適用于不同煤級的多相水精確評估方法,主要結論如下:
(1)核磁共振-離心實驗測得吸附水與自由水含量分別為15.89-39.27 mg/g與4.26-24.82 mg/g。自由水含量與離心壓力的關聯規律符合類朗繆爾方程。因實驗室離心壓力無法達到理想極限值,實驗測試值與理論計算結果存在系統性偏差。
(2)通過融合吸附比例方程與典型實驗數據(離心、比表面積及孔體積),計算出煤體吸附水平均密度約為1.59 g/cm3,平均吸附層厚度約為0.63 nm。基于最大相似性理論反演獲得最優表面弛豫率分布于2.2-5.2 nm/ms區間。
(3)采用本文提出的多相水表征方法,重新評估了飽和狀態下吸附/自由相水的理論核磁共振T2分布,發現其與實驗結果存在顯著差異。驗證表明,該多相水再評估方法不僅適用于實驗室精細分析,還可推廣至礦場測井解釋,特別為現場多相水評估提供了新方案。

中尺寸核磁共振成像分析儀
如您對以上應用感興趣,歡迎咨詢:15618820062
參考文獻
[1] Sijian Zheng, Yanbin Yao, Dameng Liu, Shuxun Sang, Shiqi Liu, Meng Wang, Xiaozhi Zhou, Ran Wang, Sijie Han. Re-evaluating the accurate multiphase water distribution in coals: Unifying experiment and theory[J]. Chemical Engineering Journal, 2023, 464, 142637.
 掃描二維碼
掃描二維碼 掃描二維碼
掃描二維碼電話:400-060-3233
售后:400-060-3233


返回頂部